The Developer's Guide to BLAST
This document is intended to explain, clarify and document the source code to NCBI's BLAST suite of sequence alignment algorithms. It is intended for developers who want to understand, modify or debug the BLAST source code. The major goals are as follows:
-
Attempt to document the "institutional memory" of the developers here at NCBI. In many cases, the algorithms used within BLAST, and the design decisions embodied in the code, have never been documented.
-
Reduce the learning curve required to become familiar with the BLAST code
-
Attempt to explain the performance characteristics of the BLAST algorithm, and to document the performance optimizations present in the code.
This document is necessarily incomplete, as BLAST is still under constant development. At best it is an aid to following a moving target. The scope of the document is limited to the 'new' BLAST engine; this is a wholesale rewrite of the original BLAST code that incorporates current algorithmic ideas for similarity searching, major improvements in readability, much more thorough documentation, and a more consistent interface for applications to use. Reference to 'the BLAST engine' or 'the BLAST code' will by default refer to this new engine; statements that apply to the original BLAST engine will be specifically labeled as such.
This section is a hideously abbreviated introduction to similarity searching, a subject whose nuances are far beyond the scope of this document. If you already know about this subject, just skip to the next section.
BLAST is designed to match up biological sequences to other biological sequences. The assumption underlying algorithms like BLAST is that biological sequences which 'look the same' perform a similar biological function and may be related through evolution. This is not always true, but is true often enough to be useful to molecular biologists.
A 'sequence' in this context is a complex organic molecule, composed of chains of atoms. Many of the biological properties of such molecules are determined by their three-dimensional shape, but comparing molecules through their shape is an inexact science. It is much simpler to assume that a molecule is a linear chain of atoms and to compare two such molecules without regard to their three-dimensional structure. Once this structure is removed, molecules can be treated as a chain of acids, and the chains can be compared.
Molecular biology assigns a symbol (or 'letter') to each acid in the chain. DNA sequences are chains of letters drawn from a 4-letter alphabet of nucleic acids ('nucleotides' or 'bases'). Protein sequences are chains of letters drawn from a 22-letter alphabet of amino acids (20 standard plus 2 unusual ones). A protein sequence can be derived from a nucleotide sequence by collecting contiguous triplets of bases together and turning the triplet into a protein letter; the mapping from nucleotide triplet to protein letter is many-to-one and controlled by a 'genetic code' (essentially a lookup table). It is often useful to compare protein sequences to nucleotide sequences: sequencing DNA is easy in this day and age, but sequencing proteins is not nearly as common. Thus, searching DNA databases for a collection of bases that happen to convert (or translate) to a known protein lets one discover proteins without having to construct them.
A list of nucleotides may be converted to a protein sequence by passing each triplet of nucleotides through the genetic code. However, the resulting protein sequence can look very different depending on the exact nucleotide where the conversion starts. Start the conversion process one nucleotide into the list and you get a protein sequence in a different 'frame'. In general, translated protein sequences only make biological sense when you guess the correct frame for translating them out of the three choices possible (i.e. 0, 1, or 2 nucleotides into the list, or into the reverse complement of the list). The translation may even have to account for a 'frame shift', where translation must switch to a different frame midway through converting the nucleotide sequence because of a sequencing error or sequence mutation. The different frames are assigned numbers by convention; the frames 1, 2, and 3 refer to converting the nucleotide sequence (the 'plus strand'), while frames -1, -2 and -3 refer to converting the reverse complement of the nucleotide sequence (the 'minus strand'). When dealing with translated sequences, BLAST works with all six translations simultaneously, so you won't have to guess which frame is biologically correct.
Protein sequences are usually small (the largest one we have is about 40000 letters, with the average length being 200-400 letters); because their composition is drawn from a large alphabet, and also because the genetic code contains redundancy which does not affect the protein sequence after translation, the amount of 'information' in a protein sequence can be high. Nucleotide sequences, on the other hand, can be extremely large (human chromosomes are strings of DNA that are hundreds of millions of bases in size) but possess low information per letter (since their alphabet is small). Large-scale studies have shown that both kinds of sequences, when derived from real organisms, are significantly different from random collections of letters. These characteristics guide the sorts of algorithms used to match up ('align') both kinds of sequences.
BLAST and many other pairwise alignment algorithms attempt to quantify the 'goodness' of a match between two sequences by assigning a score to the match. Statistical theory can be used to determine how big a score would have to be before the alignment that generated that score can be considered unlikely to have arisen by chance. Scores are calculated by assigning a score to each letter of one sequence based on the letter of the other sequence to which it aligns, then adding up all of these pairwise scores. An alignment is 'global' if it includes all the letters of both sequences, and is 'local' if it includes only a subset of one or both sequences. Less common are 'semi-global' alignments, for example aligning ungapped regions of a protein domain globally to a protein sequence. Most of the interest today is in local alignments (which BLAST finds); they let researchers isolate particular pieces of a sequence as interesting. The statistics behind local alignments are also more straightforward. A local alignment is 'optimal' if making any change to the alignment (changing its size, adding or moving gaps) lowers its score.
An alignment is 'ungapped' if gaps are not allowed inside the alignment. Allowing gaps within the alignment makes the alignment process much harder, but allows finding alignments that could never be found with ungapped methods. For example, this alignment:
tggggtc----gca
||||||| |||
tggggtcggttgca
can manage more exact matches because of the gap in the first sequence. This gapped alignment can achieve a higher score than an optimal ungapped alignment, depending on how much of a penalty is associated with a gap, and how much of a bonus is associated with matching two letters.
The job of sequence alignment algorithms like BLAST is to find most (preferably all) of the highest-scoring (disjoint, or at least minimally overlapping) local alignments between a query and one or more database ('subject') sequences. The cutoff score below which alignments are excluded from the BLAST output depends on statistical considerations described elsewhere. It is often true that there are many different-looking alignments in a given query-subject sequence neighborhood that happen to have the same score; BLAST is considered to work correctly if enough alignments are found to cover the extent of the query and subject sequences in this neighborhood. Often, the existence of an alignment between two sequences matters more than the exact form of that alignment.
BLAST is actually a collection of algorithms, each tailored to compute different kinds of local alignments (see the O'Reilly book in the Bibliography for much more information about using BLAST):
-
BLASTP aligns one or more protein query sequences to one or more subject protein sequences. The subject sequences can be grouped into a BLAST database using NCBI software tools like formatdb, or merely assembled into a text file.
-
RPSBLAST aligns one or more protein query sequences to a database of protein sequence profiles. In its present incarnation, each profile in the database is a position-specific score matrix derived from a simultaneous alignment of protein sequences that have functionally similar pieces. Rpsblast is used to detect whether portions of the query sequence are likely to perform a known biological function.
-
PSIBLAST aligns a protein query to a protein database, but attempts to build up a query-specific scoring model that will be sensitive only to database sequences that lie within the same 'protein family'. The scoring model scoring model is constructed iteratively and is embodied in a position-specific scoring matrix that takes the place of BLAST's generic score matrices.
-
PHIBLAST computes local alignments between a single protein sequence and a database of protein sequences. Unlike ordinary blastp, which works strictly on sequence similarity between query and database sequences, the local alignments produced by phiblast begin with a collection of matches, on the query and database sequences, to a specified regular expression pattern. Using a properly chosen regular expression makes this blast variant much more specific than blastp, since there are human-curated databases of protein functional sites that are encoded as regular expressions. Alignments that include these regular expressions may not have enough sequence similarity to be noticed by ordinary blastp.
-
BLASTN aligns one or more nucleotide query sequences to one or more nucleotide subject sequences. Megablast is a subspecies of blastn that is optimized for finding very similar alignments in very large sequences. In the old BLAST engine, blastn searches were handled very differently from megablast searches. The current BLAST engine has combined elements of the two types of searches together and will internally switch back and forth between them in order to compute nucleotide alignments as quickly as possible.
-
BLASTX takes as input a collection of nucleotide sequences, translates each sequence into its six hypothetical reading frames to produce six different hypothetical protein sequences, then aligns the collection of protein sequences against a collection of one or more subject sequences. Blastx is useful for tasks like determining if a newly discovered nucleotide sequence could contain a known gene within it
-
TBLASTN takes as input a collection of protein query sequences and aligns against a collection of nucleotide subject sequences, after first translating each nucleotide sequence into its six hypothetical protein sequences. Tblastn is useful for tasks like searching a newly sequenced genome for known genes.
-
TBLASTX searches a translated collection of nucleotide queries against a translated collection of nucleotide subjects. Unlike the other translated search types, it's not obvious what sorts of jobs tblastx is useful for performing. One use appearing in the literature is to align hypothetical protein sequences whose underlying nucleotides have diverged significantly from each other
There is a distinction between the format of subject sequences suitable for input to the blast engine, and the format for subject sequence suitable for blast applications. For example, the blast engine is flexible enough to work with any collection of subject sequences that the corresponding toolkit API layer supports, while the C toolkit blastall application may only be given subject sequences collected into a blast database.
The translated searches assume that each alignment found lie completely on a single translation of the converted sequence. However, the engine also allows blastx and tblastn searches to compute out-of-frame (OOF) alignments. This option finds local alignments that can slip from one translated reading frame to another in the course of an alignment. An OOF translated search is useful for recovering alignments that contain an embedded frame shift.
All of the above BLAST programs are unified into a single codebase. This simplifies the interface into the BLAST engine, and allows improvements in one low-level component to automatically propagate to all BLAST programs that use that component. The disadvantage to this approach is that portions of the engine must execute conditionally, depending on the BLAST program in use.
The NCBI C and C++ Toolkits both contain source code for the current BLAST engine. The source code is divided into toolkit-specific and toolkit-independent directories, and the latter is C code that is mirrored between the two toolkits (with the minor exception of slightly different header files to establish basic data types and such). A schematic breakdown of the engine source is as follows:
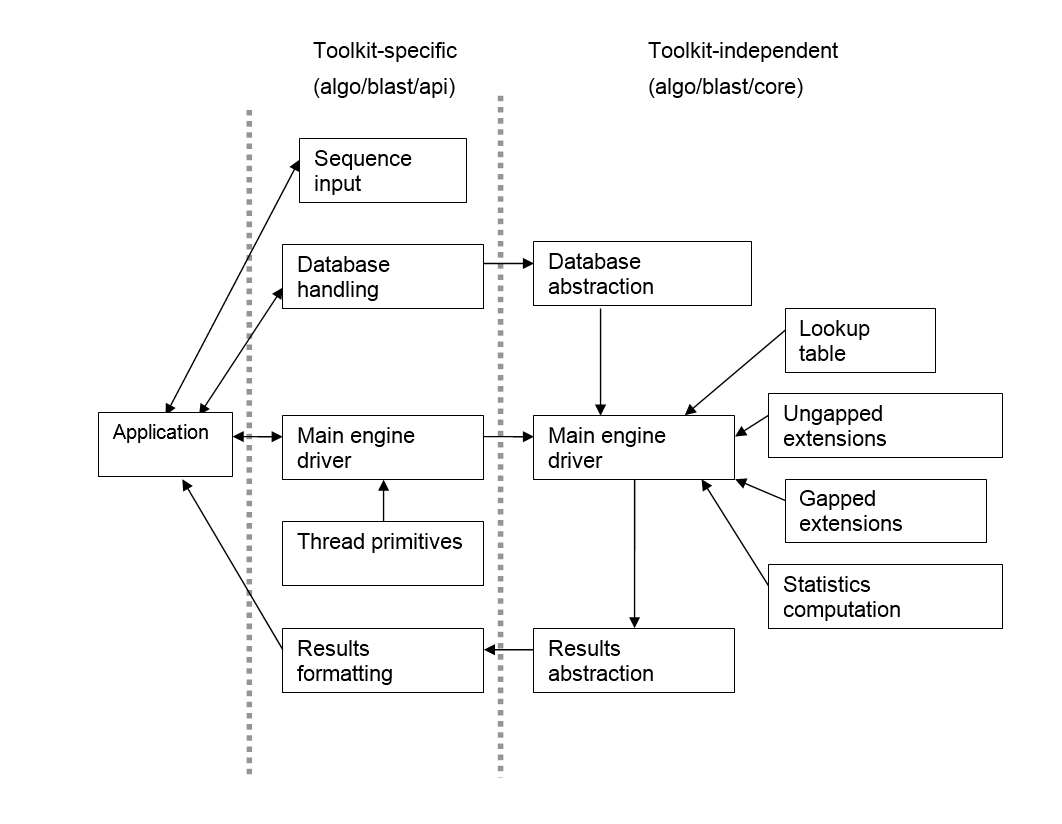
A blast search is divided into three general phases, the seed collection, ungapped extension, and gapped extension phases. The gapped extension phase is further divided into score-only and score-with-traceback extension phases. The following sections cover each of these phases in turn, then describe the construction of the main engine driver and the toolkit-specific BLAST API layout.